What is the function of the CFTR gene

Olivia Owen
Published Apr 19, 2026
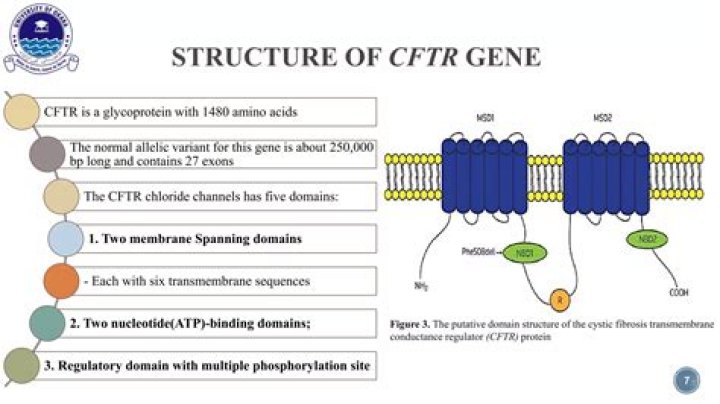
The CFTR protein The cystic fibrosis transmembrane conductance regulator (CFTR) protein is responsible for regulating the proper flow of chloride and sodium (a component of salt) in and out of the cell membranes in the lungs and other organs. is made up of 1,480 amino acids.
What type of protein is CFTR?
The cystic fibrosis transmembrane conductance regulator (CFTR) is responsible for the disease cystic fibrosis (CF). It is a membrane protein belonging to the ABC transporter family functioning as a chloride/anion channel in epithelial cells around the body.
Where is the CFTR protein in the body?
Normal function CFTR is complex protein found on the surface membrane of cells in a wide variety of tissues where it functions as a regulated chloride ion channel. In the lung CFTR is found on the apical membrane of the cells lining the airways.
How does CFTR cause cystic fibrosis?
Mutations in the CFTR gene disrupt the function of the chloride channels, preventing them from regulating the flow of chloride ions and water across cell membranes. As a result, cells that line the passageways of the lungs, pancreas, and other organs produce mucus that is unusually thick and sticky.What chromosome is CFTR gene on?
Although there have been numerous reports from around the world of mutations in the gene of chromosome 7 known as CFTR (cystic fibrosis transmembrane conductance regulator), little attention has been given to integrating these mutant alleles into a global understanding of the population molecular genetics associated …
How is CFTR regulated?
Cystic fibrosis transmembrane conductance regulator (CFTR) chloride channels are regulated tightly by protein kinases and phosphatases. The regulatory domain of CFTR has about 20 potential sites for phosphorylation by protein kinases A (PKA) and C (PKC).
How was the CFTR gene discovered?
The Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) gene was identified in 1989 by geneticist Lap-Chee Tsui and his research team as the gene associated with cystic fibrosis (CF). Tsui’s research pinpointed the gene, some mutations to which cause CF, and it revealed the underlying disease mechanism.
Which tissue S in CF patients need functional CFTR genes?
CFTR is found to be expressed in the epithelial cells of a variety of tissues and organs, whose functions are significantly affected in CF patients: lung and trachea, pancreas, liver, intestines, and sweat glands.Does everyone have the CFTR gene?
Every person has two copies of the cystic fibrosis transmembrane conductance regulator (CFTR) gene. A person must inherit two copies of the CFTR gene that contain mutations — one copy from each parent — to have cystic fibrosis.
What is the cause of his CFTR dysfunction?CF is a multiorgan genetic disease caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene and is characterized by progressive chronic obstructive lung disease. Most cases of COPD are a result of noxious particles, mainly cigarette smoke but also other environmental pollutants.
Article first time published onWhen was the CFTR gene discovered?
Mutations in a single gene – the Cystic Fibrosis Transmembrane Regulator (CFTR) gene – causes CF. The gene was discovered in 1989. Since then, more than 900 mutations of this single gene have been identified.
What is the most common CFTR mutation?
The most common CF mutation, F508del, is primarily considered to be a processing mutation. The F508del mutation removes a single amino acid from the CFTR protein. Without this building block, the CFTR protein cannot stay in the correct 3-D shape.
Is CFTR an integral or peripheral plasma membrane protein?
CFTR is an integral membrane protein and is made of a single polypeptide chain of 1480 amino acids.
Is the CFTR protein active transport?
Among human ABC proteins, CFTR is thought to be unique in that it has no active transport function, but instead acts as a phosphorylation-regulated, ATP-gated anion channel [5. The ABC protein turned chloride channel whose failure causes cystic fibrosis.
How does CFTR channel work?
In the lung, the CFTR ion channel moves chloride ions from inside the cell to outside the cell. To get out of the cell, the chloride ions move through the center of the tube formed by the CFTR protein. Once the chloride ions are outside the cell, they attract a layer of water.
What activates CFTR?
CFTR is an ATP-dependent membrane transporter which is activated by directly binding to ATP. Opening of the CFTR is initiated by ATP binding at the NBD2 site of this channel [5], [6].
How are the different CFTR mutations classified?
BIOLOGY OF CFTR MUTATION: TRADITIONAL CLASSIFICATION Traditional classification of CF mutations based on their cellular phenotype. Class I: protein synthesis defect; class II: maturation defect; class III: gating defect; class IV: conductance defect; class V: reduced quantity; and class VI: reduced stability.
Is the CFTR gene dominant or recessive?
CF is caused by mutations in the CFTR gene and inheritance is autosomal recessive .
What happens when the CFTR protein is mutated?
Mutations in the CFTR gene disrupt the function of the chloride channel, preventing the usual flow of chloride ions and water into and out of cells. As a result, cells in the male genital tract produce mucus that is abnormally thick and sticky.
Can a woman with CF have a baby?
Pregnancy is possible for women with cystic fibrosis but it can pose serious risks and challenges. If you have cystic fibrosis, it is best to visit with your health care provider to assess your personal risks before becoming pregnant.
How does cholera affect CFTR?
In the presence of cholera toxin, chloride transport into the intestinal lumen through CFTRs located on the luminal surface of intestinal crypt cells is continuously activated by intracellular cAMP causing osmotic diarrhea drawing water into the lumen and leading to the characteristic large volumes of watery diarrhea.
How many exons does the CFTR gene have?
The cystic fibrosis transmembrane conductance regulator (CFTR, OMIM: 602421) gene compromises 27 exons, is localized at 7q31. 2, and biallelic mutations lead to the CF phenotype or CAVD. To date, more than 2000 different mutations have been reported in the CFTR gene.
Which type of mutation has no effect on phenotype?
Silent mutations are mutations in DNA that do not have an observable effect on the organism’s phenotype. They are a specific type of neutral mutation.
How many alleles are there for the cystic fibrosis gene?
CFTR functions principally as a cAMP-induced chloride channel and appears capable of regulating other ion channels. Besides the most common mutation, DeltaF508, accounting for about 70% of CF chromosomes worldwide, more than 850 mutant alleles have been reported to the CF Genetic Analysis Consortium.
Why do patients with CF have salty sweat?
Why do people with CF have salty skin? In people with CF there is a problem in the transport of chloride across cell membranes. This causes thicker, stickier mucus in the lungs and digestive system, but also results in higher levels of chloride (as salt) in sweat compared with those who do not have cystic fibrosis.
What is CFTR modulator?
Cystic fibrosis transmembrane conductance regulator (CFTR) modulators are a class of drugs that act by improving production, intracellular processing, and/or function of the defective CFTR protein.
Why does CFTR use ATP?
Like other ABC transporters, CFTR uses ATP binding to its two nucleotide binding domains (NBDs) to drive conformational rearrangements of its transmembrane domains (2, 3).
Is CFTR simple diffusion?
On the other hand, the CFTR channel, although equipped with all molecular components of ABC family members, catalyzes downhill diffusion of chloride ions. For such a passive transport system, there is no obvious need for external energy input (e.g., ATP hydrolysis).
Is CFTR a channel or transporter?
Cystic fibrosis transmembrane conductance regulator (CFTR) is an ATP-gated anion channel with two remarkable distinctions. First, it is the only ATP-binding cassette (ABC) transporter that is known to be an ion channel—almost all others function as transport ATPases.